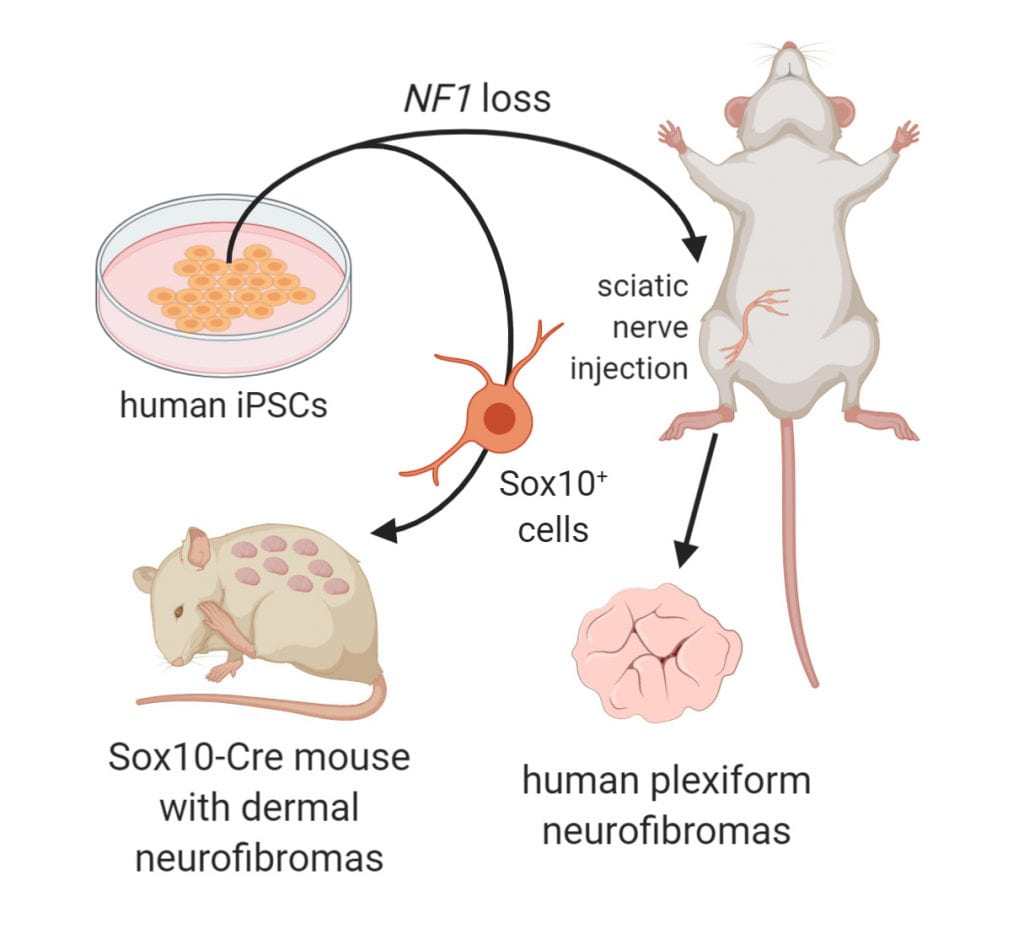
Neurofibromatosis type 1 (NF1) is a common tumor predisposition syndrome caused by NF1 gene mutation, in which affected individuals develop Schwann cell lineage peripheral nerve sheath tumors (neurofibromas). To investigate human neurofibroma pathogenesis, the Gutmann laboratory in collaboration with Dr. Lu Le (University of Texas, Southwestern) differentiated a series of isogenic, patient-specific NF1-mutant human induced pluripotent stem cells (hiPSCs) into Schwannian lineage cells (SLCs). We found that, although control and heterozygous NF1-mutant hiPSCs-SLCs did not form tumors following mouse sciatic nerve implantation, NF1-null SLCs formed bona fide neurofibromas with high levels of SOX10 expression. To confirm that SOX10+ SLCs contained the cells of origin for neurofibromas, both Nf1 genes were inactivated in mouse Sox10+ cells, leading to classic nodular cutaneous and plexiform neurofibroma formation that completely recapitulated their human counterparts. Moreover, we discovered that NF1 loss impaired Schwann cell differentiation by inducing a persistent stem-like state to expand the pool of progenitors required to initiate tumor formation, indicating that, in addition to regulating MAPK-mediated cell growth, NF1 loss also altered Schwann cell differentiation to promote neurofibroma development. Taken together, our laboratories collectively established a complementary humanized neurofibroma explant and, to our knowledge, first-in-kind genetically engineered nodular cutaneous neurofibroma mouse models that delineate neurofibroma pathogenesis amenable to future therapeutic target discovery and evaluation.
Learn more about “Modeling Human Neurologic Disease”

Yuan Mo, PhD 
Corina Anastasaki, PhD 
Jason Papke, MS
Mo J, Anastasaki C, Chen Z, Shipman T, Papke J, Yin K, Gutmann DH, Le LQ. Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. J Clin Invest. 2021 Jan 4;131(1):e139807. doi: 10.1172/JCI139807. PMID: 33108355; PMCID: PMC7773354.